In this article, we have made a comprehensive overview of research findings on the immune system in patients with myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). In contrast to what is frequently claimed, current data do not suggest that (low-grade) inflammation is a key driver of ME/CFS symptoms. There have been major studies into viral persistence, antibodies, and cytokines, but with mostly null results. The most consistent findings in the ME/CFS literature on the immune system over the past 40 years are reduced natural killer cell cytotoxicity and increased levels of the signaling molecule TGF-beta.
We have divided this overview into separate chapters, so you can read one at a time and come back to read the rest later. There are chapters on:
– viral persistence
– cytokines
– neuroinflammation
– autoantibodies
– immune cells such as Natural Killer cells, T-cells, and B-cells.
Although we took great care in dissecting and interpreting research findings, we do not have a professional background in medicine or immunology. We are therefore interested in hearing comments and feedback from professionals in the field. If you think we missed an important study or replicated finding, feel free to post them in the comments section below.
Introduction
Viral triggers
Several studies have shown that the incidence of ME/CFS is increased after infectious diseases such as mononucleosis, COVID-19, or Q-fever. This is how the syndrome was first recognized in the past century, following viral outbreaks and young adults with Epstein-Barr virus infection. In the past years, the SARS-CoV-2 pandemic confirmed that certain infections are indeed a risk factor for developing ME/CFS.
This observation has made the immune system a key target in studying the pathophysiology of ME/CFS. Popular hypotheses for explaining the syndrome’s long-term symptoms include an inability to clear pathogens, misdirected antibodies, and an inflammatory response that is never fully resolved after the acute infection. All three have been thoroughly studied but mostly given null results.
An echo or side-effect
Although countless papers contend that (low-grade) inflammation is driving symptoms in ME/CFS, the data do not point in that direction. There’s a contradiction between what papers claim in this regard and what the actual evidence shows. We believe these negative results should receive more recognition as they could help us understand the pathophysiology of ME/CFS. Inflammatory responses are ubiquitous in medicine. They are not only important in autoimmune diseases or in fighting off pathogens, but also in cancer and cardiovascular disease. Yet, the inflammatory response we see in ME/CFS is weak to non-existent. Its size contrasts with the enormous disability the illness causes.
This suggests that what’s driving symptoms is either hidden or involves a different immune pathway that we don’t fully understand yet. The most consistent immune findings in ME/CFS, such as reduced natural killer function or increased TGF-beta cytokines, are likely just an echo or side-effect of the core pathology. Rather than clinging to an inflammatory model of ME/CFS, we hope that future theories will account for the observation that we see surprisingly little immune activation for a debilitating disease triggered by viral infections.
Before we move on to discussing the evidence, beware that there will likely be papers that contradict our claims. There are numerous small studies published in obscure journals that were never replicated, supporting almost any theory you can think of. Our goal, however, is to see the forest through the trees. That’s why we focus on big and high-quality studies and findings that were replicated by multiple research teams.
Viral persistence
Dead ends
Let’s start with viral persistence, the idea that a virus is (still) inside ME/CFS patients and causing their symptoms. This has been one of the most prominent lines of research in ME/CFS, especially in the 1980s and 1990s, when it drew inspiration from the fight against HIV and AIDS.
There have been investigations into enteroviruses (by the Scottish team of Peter Behan), human herpesvirus 6 (by co-discoverer Dharam Ablashi and Bhupesh Prusty), the Epstein-Barr virus (by Stephen Straus and Jay Levy), retroviruses (by Elaine DeFreitas and Judy Mikovitz), and Parvovirus B19 (by Jonathan Kerr). Smaller studies looked at a potential link between ME/CFS and cytomegalovirus, chlamydia pneumonia, and hepatitis C. But almost all of these enquiries led to a dead end.
Lipkin’s null results
The biggest study on viral persistence was done by the well-known ‘virus hunter’ Ian Lipkin at Columbia University. Lipkin’s team screened the blood, saliva, and feces from ME/CFS patients recruited from five different clinical sites. They looked for traces of a long list of viruses but found no significant differences compared to controls. The authors concluded that there is likely little to be found: “Our findings suggest that future investigations into viral infections in ME/CFS should focus on adaptive immune responses rather than surveillance for viral gene products.”
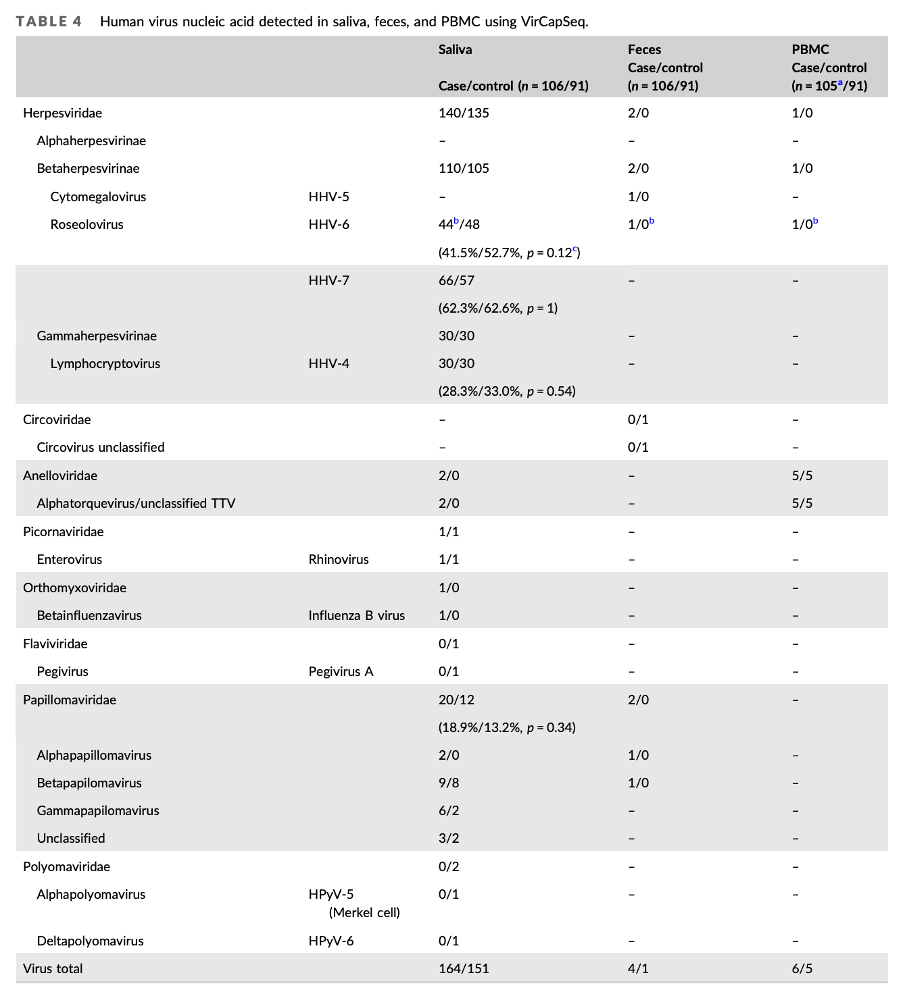
A study on twin pairs from the University of Washington found similar results. The team of Ronald Davis at Stanford University screened 185 viruses in the blood of severe ME/CFS patients, but “surprisingly, more viruses were found in the healthy controls than in the ME/CFS patients.” Akiko Iwasaki at Yale used antibody repertoires to determine past exposures to pathogens, but also found nothing remarkable in the Swedish cohort they tested. Iwijn De Vlaminck’s lab at Cornell University measured cell-free RNA that had leaked out from different cells and tissues into the blood. If there’s a virus hiding somewhere in the body, this free RNA could reveal its presence, but unfortunately, no such clues were found. The CureME biobank also found no difference in the seroprevalence for six herpes viruses.
EBV-reactivation
Reactivation of the Epstein-Barr Virus (EBV) is also a popular hypothesis associated with ME/CFS. A prominent German study, however, measured antibody responses against 3054 peptides associated with EBV. Unlike the results seen in patients with multiple sclerosis and lupus, those with ME/CFS had a response similar to healthy controls. Data from the UK Biobank also showed that having persistent DNA of EBV in blood is not associated with (self-reported) ME/CFS, while it is linked to several autoimmune diseases.
Researchers from Ohio State University did report that ME/CFS patients have increased antibodies against dUTPase, an enzyme EBV uses to replicate its DNA, but these findings haven’t been confirmed by others.
Chia’s biopsies
A 2007 study by Dr. John Chia deserves special mention as it found signs of enterovirus in stomach biopsies from 165 ME/CFS patients. There are, however, several reasons to be skeptical about this finding. Most of the virus samples could not be cultured, there was no cytopathic effect (no evidence that it was causing cell damage), and the findings were never replicated. The ME/CFS community already has two bad experiences with research on retroviruses such as XMRV and HTLV, which announced major findings but eventually turned out to be a fluke. Dr. Chia isn’t an academic researcher and had previously claimed to have found elevated antibodies against enteroviruses in blood, a finding that is contradicted by other research.
Treatment trials
Lastly, several small randomized trials of antiviral treatments, such as acyclovir, valganciclovir, Isoprinosine, and alpha interferon, have shown no effect in ME/CFS. The findings on Ampligen (rintatolimod) are more interesting. The drug acts as a TLR-3 agonist, thereby activating a major antiviral pathway in the immune system. A large study of Ampligen found an improvement in exercise duration in ME/CFS patients, but the effect on symptoms and functional ability was less impressive. The Food and Drug Administration (FDA) also saw problems in how the company Hemispherx Biopharma changed its analysis plan during the trial. It was not convinced of the effectiveness of Ampligen and rejected its approval in the US.
Conclusion
In conclusion, there’s mostly negative data regarding viral persistence in ME/CFS. One limitation is that few studies have looked at tissue biopsies, something that multiple Polybio-funded studies hope to address. If a virus is key to ME/CFS, however, it appears to be exceptionally good at hiding itself while causing chronic, debilitating symptoms. It seems to cause no obvious tissue damage, leave no traces in blood, feces, or saliva, and induce no systemic immune activation.

Cytokines
That last point brings us to our second topic: inflammation and markers of immune activation.
Classical inflammation
Historically, inflammation is defined by four cardinal signs: redness, heat, pain, and swelling (in Latin: rubor, calor, dolor, tumor). These occur when blood vessels widen and become leaky, allowing immune cells to enter tissue and fight a perceived threat. While this is the primary mechanism of diseases like rheumatoid arthritis, it’s not a typical feature of ME/CFS. Standard markers of inflammation, such as CRP (C-Reactive Protein) and ESR (erythrocyte sedimentation rate), are normal in most patients.
No consistency
A more refined hypothesis, therefore, argues that ME/CFS is not driven by classical inflammation but by a more subtle immune activation smoldering in the background. This can be tested by measuring cytokines, the small signaling molecules of our immune system. Typical inflammatory cytokines are, for example, TNF-alpha, IL-6, and IL-1. More than thirty studies have tested cytokines in the blood of ME/CFS patients, and unfortunately, the results have largely been inconsistent (see reviews here, here, and here). As one systematic review concluded: “of the 64 cytokines analysed, none appear to differ with any consistency between CFS/ME/SEID patients and healthy controls, in either serum or other physiological fluids, despite comparability between each study’s analytical methods.” The cytokine most often found to be increased in ME/CFS is TGF-β, which often acts as an anti-inflammatory signal (more about that later…).
Short versus long illness duration
Two big studies are worth zooming in on. The first, by Stanford researchers Jose Montoya and Mark Davis, tested 51 cytokines in 192 ME/CFS patients and 392 healthy controls. Although it confirmed increased levels of TGF-beta in patients, it also found that this cytokine does not correlate with ME/CFS severity. In contrast, some inflammatory cytokines, such as interferon-gamma, did correlate with severity, but their levels were similar to those of controls.
The second major study was done by Mady Hornig and colleagues at Columbia University. It had an even bigger sample size: 298 patients and 348 controls, all matched for key variables such as sex, age, location, and time of sampling. In this study, patients often had lower levels of circulating cytokines. But zooming in, the researchers found a pattern: patients with a short illness duration (≤3 years) had increased cytokine levels compared to controls, while those with long duration had lower levels. In other words, there seems to be a trajectory: early in the illness, ME/CFS patients show (slightly) elevated cytokine levels, but this immune signature disappears over time. Although this was just an exploratory observation, it fits with Long Covid studies having more success at finding elevated inflammatory markers – likely because they test patients closer to the initial immune trigger.
Post-exertional malaise
Overall, these cytokine studies suggest that the inflammatory responses we can measure in the blood are unlikely to be driving ME/CFS symptoms. There are, however, some important caveats to address.
One common criticism is that cytokines should be measured during post-exertional malaise (PEM), when patients experience a worsening of their symptoms. Several studies have tried to do this, for example, by measuring cytokines after an exercise test or other stressor such as sleep deprivation. These studies were smaller, so their results are less certain, but most also found null results. As one review concluded: “Exercise does not result in abnormally higher levels of pro- or anti-inflammatory cytokines in patients with CFS.” Another review stated that “following physical exercise, there were no differences in circulating cytokine levels between cases and controls.”
This is curious because cytokine levels can rise dramatically after certain events. In people who ran a marathon, researchers found 30-fold increases in cytokines such as IL-6. The lack of group differences after exercise makes it unlikely that such cytokine responses are driving PEM or other ME/CFS symptoms, but there are a couple of potential explanations. Perhaps the cytokines were measured too soon after exercise, while PEM only kicks in later. Or the exercise test might not have been sufficient to induce PEM in most participants. Studies with a longitudinal design and home-based testing of PEM might be able to give a clearer answer.
Cerebrospinal fluid
A second caveat is that most of these studies measured cytokines in the blood, while these signaling molecules often have a local effect only, usually near an infection or injury. What we see in the blood is likely just a spillover, a weak echo of what’s going on in tissue. There have been a couple of ME/CFS studies, however, that measured cytokines in the cerebrospinal fluid that surrounds the brain. These have also found no consistent pattern, perhaps due to their small sample sizes. Examples are available here, here, here, and here.
TGF-beta
A third limitation is that these studies are hard to do correctly, because cytokines are influenced by multiple factors such as sex, age, BMI, time of measurement, etc. It’s therefore crucial that samples from cases and controls are handled in the exact same way. One study, for example, suggested that the finding of increased TGF-beta in ME/CFS patients could be due to different centrifuge forces used for rotating the blood samples. Lower force means more platelets, which are a rich source of TGF-beta. Using another centrifuge force in patients versus controls might have caused group differences.
But suppose the TGF-β finding is real: what would it mean? The answer isn’t straightforward. TGF-β is a multifunctional cytokine whose many roles depend heavily on context. It often acts as an anti-inflammatory signal and is perhaps best known for stimulating the production of extracellular matrix proteins and tissue repair. In fibrosis and systemic sclerosis, for example, TGF-beta is known to be overactive.
Anakinra trial
When it comes to treatments, we found only one trial that targeted inflammatory cytokines. The Dutch group led by Jos Van der Meer tested Anakinra, a drug that blocks the receptor of the inflammatory cytokine IL-1 (it inhibits both IL- 1-alpha and IL-1-beta). 50 ME/CFS patients were enrolled, but Anakinra did not significantly improve their fatigue or functioning.
Conclusion
Our conclusion for immune activation and cytokines is similar to that of persistent pathogens. The current evidence for low-grade inflammation in ME/CFS is weak and not in proportion to the disability patients experience. If this mechanism is driving symptoms, it must be in a place where we can’t easily see or measure it. This is not impossible, though. The human body has immune-privileged sites where our immune system works differently. In the gut, for example, it’s allowed to be more active and aggressive to keep bacteria in check. In the brain, on the other hand, it needs to be extra cautious not to damage neurons. One popular hypothesis, therefore, argues that the inflammatory response in ME/CFS is restricted to the brain, where we can hardly see it. That’s a topic that we will tackle in our next chapter.
Neuroinflammation
In the previous chapter, we found that current evidence for (low-grade) inflammation in ME/CFS is quite limited and not in proportion to the disability patients experience. Most of these studies, however, were based on measurements in blood where immune cells are merely patrolling. Perhaps the real action is taking place in the brain, where it’s much more difficult to measure what’s going on. It would also explain why many ME/CFS symptoms are neurological in nature.
PET scans
The resident immune cells in the brain are called microglia, and one of the best ways to study them is by using PET (positron emission tomography). These PET scans inject a radioactive molecule that binds to TSPO, a protein that increases when microglia are activated. The more TSPO signal on the brain scan, the more evidence that the immune cells are activated.
Thus far, only two PET studies have been published, and both included only 9 ME/CFS patients. The first, by the Japanese group of Nakatomi et al., found increased signal in multiple brain regions of ME/CFS patients, mostly the midbrain, while the Dutch study by Raijmakers and colleagues found no significant difference with controls.
In the pipeline
Luckily, there are more of these PET studies in progress. Jarred Younger from the University of Alabama received an NIH grant to study neuroinflammation in ME/CFS. Using an improved radioactive tracer, his team has identified signs of increased microglial activation in ME/CFS patients — findings he has shared on his YouTube channel.
Michael VanElzakker from Tufts University reported similar results in Long Covid and ME/CFS patients during a conference presentation. Michelle James from Stanford shared images where it was not the brain but the muscles of ME/CFS patients that lit up on a whole-body TSPO scan.
These are just preliminary findings, though. Because of the small sample size and many brain regions studied, imaging studies like these tend to be prone to false positives. TSPO is also not exclusive to microglia. An increased signal often means something more benign than neuroinflammation, such as changes in cell metabolism, energy production, and oxidative stress. The term neuroinflammation in this context has been criticized for being a misnomer (see critiques here, here, and here). Increased TPSO signal has been reported in multiple conditions, including depression, fibromyalgia, and chronic pain.
Autopsies
There’s also evidence from 10 ME/CFS brain biopsies from the Netherlands Brain Bank, which speaks against neuroinflammation. The Dutch researchers found no evidence of microglia activation or immune cell infiltration. Instead, the microglia of ME/CFS patients looked fragmented and worn out. Similar results were found with diffusion tensor imaging, where markers of cell infiltration and edema were not increased, as you would expect with neuroinflammation, but decreased.
Indirect results
Several studies have hinted at neuroinflammation in ME/CFS using other imaging techniques. The most consistent finding is increased lactate in the ventricles, large cavities in the brain filled with cerebrospinal fluid. This has been pursued by Dikoma Shungu and colleagues at Mount Sinai using magnetic resonance spectroscopy (MRS).
Lactate accumulates when cells switch to anaerobic glycolysis, an alternative form of energy production that doesn’t need oxygen. It’s used in times of high energy need or distress. Increased lactate might therefore point to microglia activation, but it could mean numerous other things as well. The same is true for other metabolites such as glutamate or choline: they only give an indirect clue of what’s going on in the brain.
Conclusion
Overall, data on immune activation in the brain remains limited and contradictory. Current findings suggest no classical neuroinflammation with immune cell infiltration, but there may be low-level activation of microglia. Most of this story still needs to be written.
Autoantibodies
More evidence is available for another major immune hypothesis: the idea that autoantibodies cause ME/CFS. Autoantibodies are proteins that mistakenly attack the body’s own healthy cells instead of harmful invaders. An important hint in that direction is given by ME/CFS’s high female predominance. Just like many autoimmune diseases involving antibodies, ME/CFS mostly affects females.
Adrenergic and muscarinic receptors
The autoantibody hypothesis of ME/CFS received a major boost when German immunologist Carmen Scheibenbogen got involved in the field. Her team at the prestigious Charité Hospital in Berlin published a major study in 2016. It showed that ME/CFS patients have increased autoantibodies against adrenergic and muscarinic cholinergic receptors. More precisely, patients had significant elevations of antibodies against the beta-2 adrenergic (β2), M3 muscarinic acetylcholine (M3), and M4 muscarinic acetylcholine (M4) receptors.
These receptors play an important role in the autonomic nervous system, and the antibodies that target them had previously been implicated in Postural Orthostatic Tachycardia Syndrome (POTS). Researchers suspect that these antibodies contribute to orthostatic tachycardia and a range of other symptoms. Scheibenbogen’s study suggested a similar mechanism may be at work in a subset of ME/CFS patients. The evidence, however, has several major limitations.
First, there’s a lot of overlap between patients and controls. As Rheumatologist Jonathan Edwards noted on the Science for ME forum: “the difference in antibody levels between ME patients and controls is minimal and provides clear evidence for these autoantibodies NOT being a cause of symptoms.”
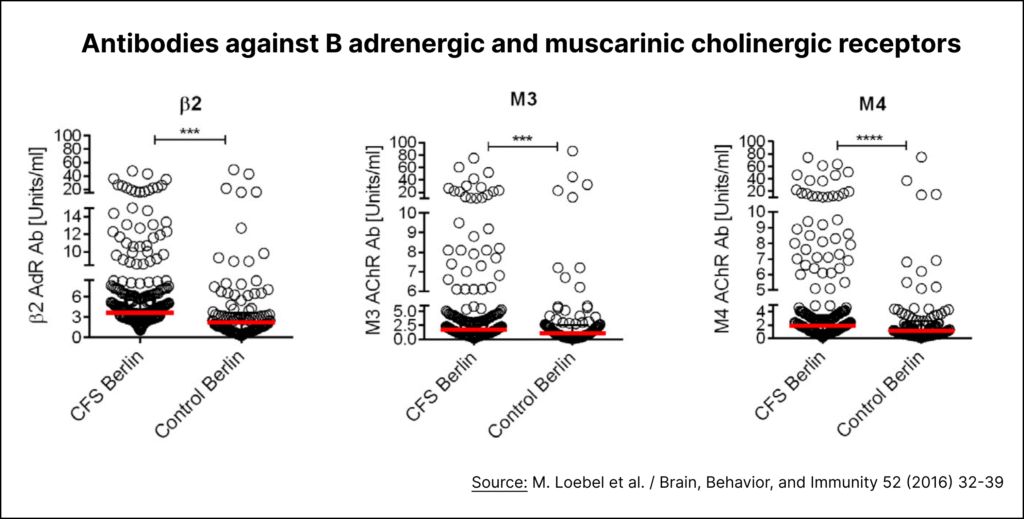
Second, the correlation between these antibodies and ME/CFS symptoms is surprisingly weak* (r < 0.3). A Swedish replication study commented: “This lack of clear correlation between antibody levels and clinical symptoms challenge the clinical relevance of autoantibodies to adrenergic and muscarinic receptors and their connection to the pathology of ME.”
Third, there have been questions about the reliability of the CellTrend assay used to measure the antibodies. A POTS study found that virtually all patients and controls had autoantibody levels against the α1 adrenergic receptor above the seropositive threshold, suggesting an error in the assay. Antibodies against β2, M3, and M4 receptors did not differentiate patients and controls.
Fourth, while some ME/CFS studies found similar results (examples here and here), others were unable to replicate the findings. In 2025, the group of Maureen Hanson at Cornell University published the most extensive study on antibodies in ME/CFS to date. It used two advanced techniques (REAP and Luminex) to test thousands of different antibodies. Despite a large sample size, it could not find any clear abnormalities. The authors write that “unlike earlier reports, our analysis of 172 participants revealed no significant differences in autoantibody reactivities between ME/CFS patients and controls, including against GPCRs such as β-adrenergic receptors.”
Rituximab and daratumumab
While measurements of antibodies have not produced strong leads, treatment trials have provided more exciting results. The Scheibenbogen group, for example, conducted a pilot study of immunoadsorption, an intervention that filters antibodies out of the blood. Some of the participants showed improvement, but it was limited and not sustained. There are plans for a randomized, sham-controlled trial on immunoadsorption in ME/CFS, so hopefully that will tell us more if this treatment works. If it does, it would put antibodies front and center in ME/CFS research.
The Rituximab trial is also an important datapoint. This cancer drug targets B-cells, the immune cells that produce antibodies. More precisely, it targets cells that have the surface marker CD20. Although initial studies reported improvement with Rituximab, a well-conducted randomized trial showed it likely does not work in ME/CFS patients.
The Norwegian researchers behind this study did not give up, though. They are now pursuing another cancer drug: daratumumab, which targets plasma cells. These are long-lived and matured cells that produce lots of antibodies but do not express the CD20 protein. They hide inside the bone marrow or gut and could have survived a short-term rituximab treatment. Instead of CD20, they express the CD38 protein on their surface, which is precisely the target of daratumumab. A pilot study showed promising results, and a randomized trial called RESETME is already being set up. Hopefully it will fare better than the one on rituximab. The Scheibenbogen group has also announced plans to target the same CD38 marker on plasma cells, probably with the drug Isatuximab.
HLA-genes
The big genetic study DecodeME also holds important information for the antibody hypothesis. There’s a specific location in our DNA on chromosome 6 that’s crucial for differentiating your own cells from foreign invaders. It encodes Human Leukocyte Antigens (HLA), proteins that make sure your immune system doesn’t attack your own cells. Pretty much all autoimmune diseases – multiple sclerosis, ankylosing spondylitis, type 1 diabetes, psoriasis, Hashimoto’s, etc. – are linked with some DNA variants at this HLA region on our genome. If ME/CFS is an autoimmune disease involving antibodies, we would expect to see a genetic hit there.
Unfortunately, the first analysis of DecodeME was ambiguous about this. It found that a particular HLA variant (HLA-DQA1*05:01) was protective against ME/CFS. But normally this variant is linked with other HLA variants, and that wasn’t the case here. So, there might have been an issue in the analysis. The HLA region is notoriously difficult to measure because many of its genes look alike. The DecodeME researchers therefore, plan to redo the HLA analysis to verify any potential association.
So are antibodies involved in ME/CFS? In the coming years, we will likely have a reasonable answer to that question. If the HLA-analysis, immunoadsorption, and daratumumab trial all come up negative, then the antibody theory would take a big hit. If any of these does show an effect, however, it would be the biggest breakthrough in ME/CFS research thus far.
Immune cells
In our last chapter, we will look at immune cells. Several studies have reported differences in the proportion of certain immune cell types in ME/CFS, but findings have been inconsistent. It’s easy to get a false positive due to group differences in lifestyle or selection bias – patients seeing an immunologist might not be representative of the ME/CFS population as a whole.
One of the biggest studies on this topic was published by the CureME biobank in the UK. It collected data on 251 ME/CFS patients and compared their immune cells to 107 healthy controls and 46 patients with Multiple Sclerosis. The proportion of immune cells in ME/CFS patients was largely similar to that of controls.
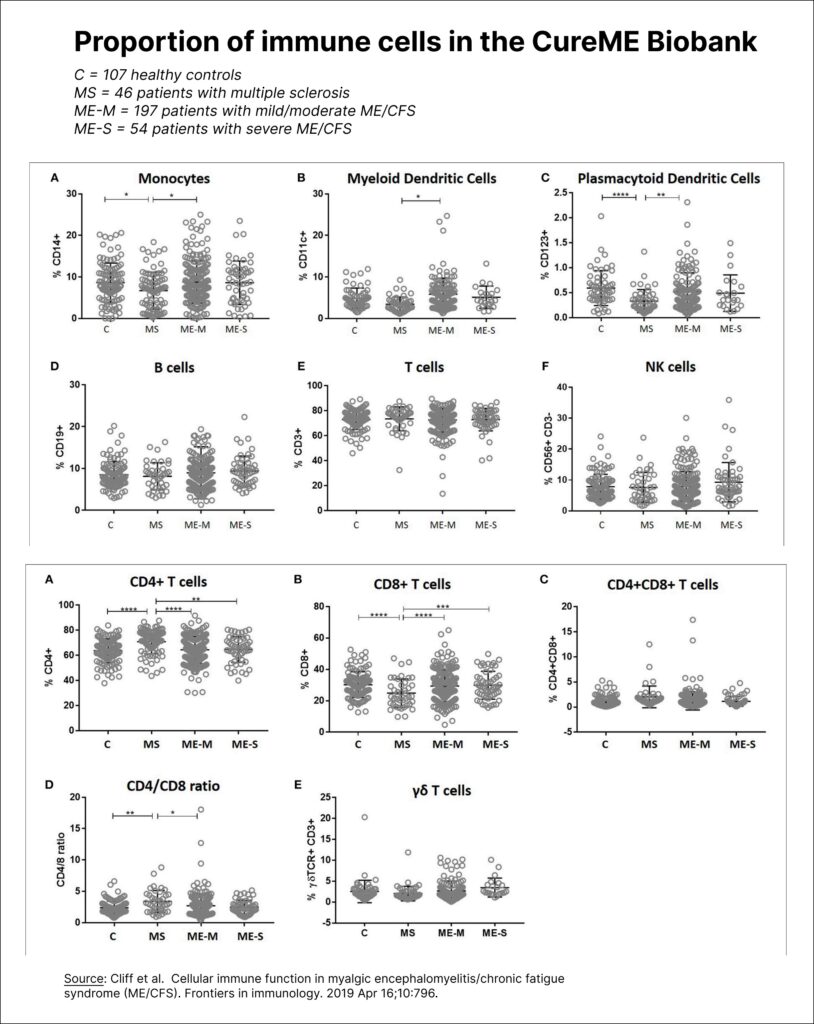
In this study, patients did have increased proportions of MAIT (mucosal-associated invariant T cells) and effector memory CD8+ T cells, but the effects were minor and haven’t been replicated.
Natural Killer cells
Although the number of Natural Killer (NK) cells seems normal, multiple studies have reported a lowered cytotoxicity in ME/CFS: a reduced ability to kill. The cells in our body are normally watched over by T-cells. These use HLA proteins to look at what’s inside the cells. If T-cells notice viral particles, they will attack and destroy the tainted cell to prevent the virus from spreading. Some viruses and cancers, however, have found a way around this by blocking the formation of HLA proteins. This prevents T-cells from checking what’s inside. In such situations, NK cells are crucial. They patrol cells to see if they have a valid HLA protein on their surface. If they don’t, then something fishy is going on, and the NK cells are there to destroy them.
Researchers test the cytotoxicity of NK cells by mixing them with cancer cells tagged with radioactive chromium. This allows them to measure the number of cancer cells killed after a couple of hours. Numerous studies have found the cytotoxicity of NK cells to be reduced in ME/CFS patients. The effect is often quite noticeable. In a big study by Nancy Klimas at the University of Miami, the median cytotoxicity value in 176 patients was less than half that of controls. The Australian team of Sonya Marshall-Gradisnik also conducted a longitudinal study with multiple measurements over time, showing that NK function was consistently decreased over a period of 12 months. These two research teams have published the most papers on the topic, but the research goes way back to the 1980s, before the name ‘chronic fatigue syndrome’ was coined. Reduced NK function was found in patients with the chronic Epstein-Barr virus and those who got ill during the mysterious outbreak at Lake Tahoe.
Of the more than 30 studies that have tested NK cytotoxicity, most have found reduced values in ME/CFS patients, but there are some exceptions. The large CureME biobank cohort and an in-depth study on patients from Oslo and Stockholm found no effect, possibly because they used frozen samples. More difficult to explain are the negative results of the multi‑site clinical assessment of ME/CFS (MCAM) study. This was a big American study organized by the Centers for Disease Control and Prevention (CDC). 174 ME/CFS patients were recruited at five specialized clinics. Samples were shipped overnight to the same laboratory for analysis. The study found a large variation in percent cytotoxicity with no difference between patients and controls. The CDC researchers used a different, more modern approach to count the number of killed cancer cells (using fluorescence instead of chromium release). Together with the overnight shipping, this made their methods a bit different from those of previous studies. The CDC, however, compared their approach to the gold standard and found that it gave similar results (R^2 = 0.84).
Unfortunately, low NK cytotoxicity isn’t a very specific finding. It has been reported in numerous conditions such as endometriosis, depression, cancer, and rheumatoid arthritis. Factors such as stress, reduced sleep, and smoking have been found to reduce NK cell function.
It’s also unknown what causes the reduced killing ability in ME/CFS. The team of Marshall-Gradisnik thinks it’s due to a dysfunction of ion channels in NK cells, but there are many other possibilities. An attractive explanation is TGF-beta, the cytokine most consistently found to be increased in ME/CFS, and also a potent inhibitor of NK cell cytotoxicity.
Treatments that increase NK-cell function, such as BioBran, Isoprinosine, and interferon-alpha, did not lead to an improvement in ME/CFS patients.
T-cells
While some older studies reported lowered T-cell proliferation in ME/CFS, findings have been less consistent than those on NK cell function. The extensive study on patients from Oslo and Stockholm found no abnormalities in cytotoxic T-cells, but it used frozen samples in which some information may have been lost.
In 2017, there was excitement as Mark Davis from Stanford reported ‘clonal expansion’ in ME/CFS during a conference presentation. Each T-cell has its own specific receptor for attaching to a specific pathogen. If they find a target that matches their receptor, they start making copies or ‘clones’ of themselves. By reading out all the T-cell receptors in an individual, researchers can check if clones have been made. It’s a major indication of immune activation, but unfortunately, it didn’t pan out in ME/CFS. Mark Davis never formally published his findings, and a replication study from Chris Ponting’s group in Scotland found no indication of clonal expansion in ME/CFS.
The T-cell abnormalities that have been reported in ME/CFS are more subtle. One of the most interesting studies came from Maureen Hanson’s group at Cornell. It found that the T-cells of ME/CFS patients have normal mitochondrial respiration but reduced glycolysis. Mitochondrial respiration is the form of energy production that uses oxygen and is highly efficient. Glycolysis is less efficient but doesn’t need oxygen and is much faster at generating energy. It’s what T-cells use to switch to activation mode. The results by Hanson’s group might mean that ME/CFS cells have more trouble getting activated or that they have a different metabolism. There are also some smaller studies suggesting immune cells of ME/CFS patients have a higher consumption of alternative fuels (amino acids or lipids) to produce energy, but these findings are all preliminary.
B-cells
This brings us to B-cells, the immune cells that produce antibodies. These have been studied in ME/CFS patients by immunologists Amolak Bansal and Geraldine Cambridge, but there wasn’t much that stood out.
The most consistent finding thus far is a skew in the B-cell receptor repertoire. A B-cell receptor is an antibody that is still attached to the surface of the cell and is used to recognize pathogens. Our immune system needs an almost endless diversity of these receptors so that it can respond to all viruses, fungi, and bacteria. One way it does this is by selecting and rearranging the building blocks for antibodies in new B-cells, a bit like shuffling cards. IGHV3-30 is one of these building blocks. Researchers have found that B-cells of ME/CFS patients used it more often in their receptors than controls.
This was reported by a Japanese group and Ian Lipkin’s team and was recently replicated by a third team from the University of Edinburgh, led by Audrey Ryback. A skewing of the B cell repertoire is normally a sign of antibody production or an immune response to a specific infection. But in these scenarios, there would also be signs of B-cells copying themselves and refining their antibodies, which Ryback couldn’t find in ME/CFS patients. It’s therefore a bit of a mystery what it means. Some selection process must have favored this building block or made it more useful in ME/CFS patients than in controls. One caveat, however, is that the replication study only found IGHV3-30 to be more common in mild and moderate ME/CFS patients, not in those with severe ME/CFS.
General conclusion
Several findings didn’t easily fit our overview. For example, two studies (available here and here) have reported an increased mannose-binding lectin deficiency in ME/CFS. This simply could be the result of selection bias, but perhaps it’s another pieces of the puzzle.
Overall, however, immune studies in ME/CFS have failed to provide strong leads that might explain the syndrome’s debilitating symptoms. There have been well-sized studies into viral persistence, antibodies, and cytokines that produced null results. There is very little evidence for the popular hypothesis of low-grade inflammation.
This has important implications for future research. Immune hypotheses on ME/CFS will need to explain and account for these negative findings.
Research should ideally focus on innovative methods targeting the gut, brain, or other tissues that have been difficult to sample. We’re looking for an immune signal that is potent enough to produce extremely debilitating symptoms yet produces no systemic immune activation visible in blood. The answer to this riddle may have implications in medicine that go far beyond ME/CFS.
Notes
* The Freitag et al. 2021 study reported that antibodies against beta-adrenergic and muscarinic acetylcholine receptors correlate with symptom severity and disability. But looking at the data in the supplementary file, Table S3, the correlation was quite weak to nonexistent, even in the subgroup with infectious onset. None were significant after applying the Benjamini-Hochberg correction for multiple comparisons.
Are there any studies on phages? (Virophage, virus killers) ?
Thank you for doing this! Recently, I’ve seen articles documenting long covid cytokine levels that are BELOW normal range, as opposed to high. I wonder if the ME/CFS research also needs to evolve to look at below range cytokine levels instead of only focusing on raised cytokine levels. Seems like the research/testing is lacking in this regard. I saw a recent Labcorp cytokine panel test result and if the result was below the normal range, the results did not flag it at all and, for the most part, it also did not measure a specific number but only reported it as “less than” the normal range. Seems like labs and researchers need to start considering below range cytokine levels and whether this has any meaning for ME/CFS or otherwise.
Hi, I’m commenting here because the `CONTACT` section of this website doesn’t seem to work and is empty.
I deeply appreciate your work here and would love if you could take a look at theory of blood clots, microthrombi and hypercoagulability – especially around the University of Cape Town.
Thanks for any comments or replies and keep up the great work!
Hi Michael thanks. Apologies about the contact section, hope it is fixed now. I’ll add the microclot theory to my list of subjects to delve into but it might take a while because there are several others topics I haven’t managed to do yet.
I have the impression that the popularity of the theory has declined quite a bit in recent years. Been a while since a saw a prominent publication on it.